1. Computational analysis of bacterial genomes and prediction of antimicrobial resistance. We develop computational tools for the automated analysis of bacterial genome sequences. This includes:
* Genome assembly
* Detection and removal of contamination by other species
* Prediction of MLST type
* Construction of phylogenetic trees from core genome sequences
* Identification of known resistance genes
* Analysis of the genomic context of resistance genes
* Detection and characterisation of plasmid sequences.
Several tools for these analyses have been developed by our group, including StrainSeeker (Roosaare et al., 2017), PlasmidSeeker (Roosaare et al., 2018) and PhenotypeSeeker (Aun et al., 2018).
Large-scale bacterial genome analysis can serve different purposes. The most common aim is the epidemiological analysis of the spread of bacterial strains and/or their resistance genes (Telling et al., 2018; Bilozor et al., 2019; Sepp et al., 2019; Telling et al., 2020).
Another frequent goal is functional analysis of genomic variants that emerge under strong natural selection in specific environment. For example, Jõers et al., 2019 describe genes mutated in a strain that is less responsive to muropeptides, while Aun et al., 2021 identified genes responsible for nodule colonization in Sinorhizobium meliloti.
Currently, our group is actively working towards developing statistical prediction models to predict virulence, antimicrobial resistance or other properties of bacterial strains based on their genomic sequences. To achieve this, we combine machine learning methods with biological knowledge.
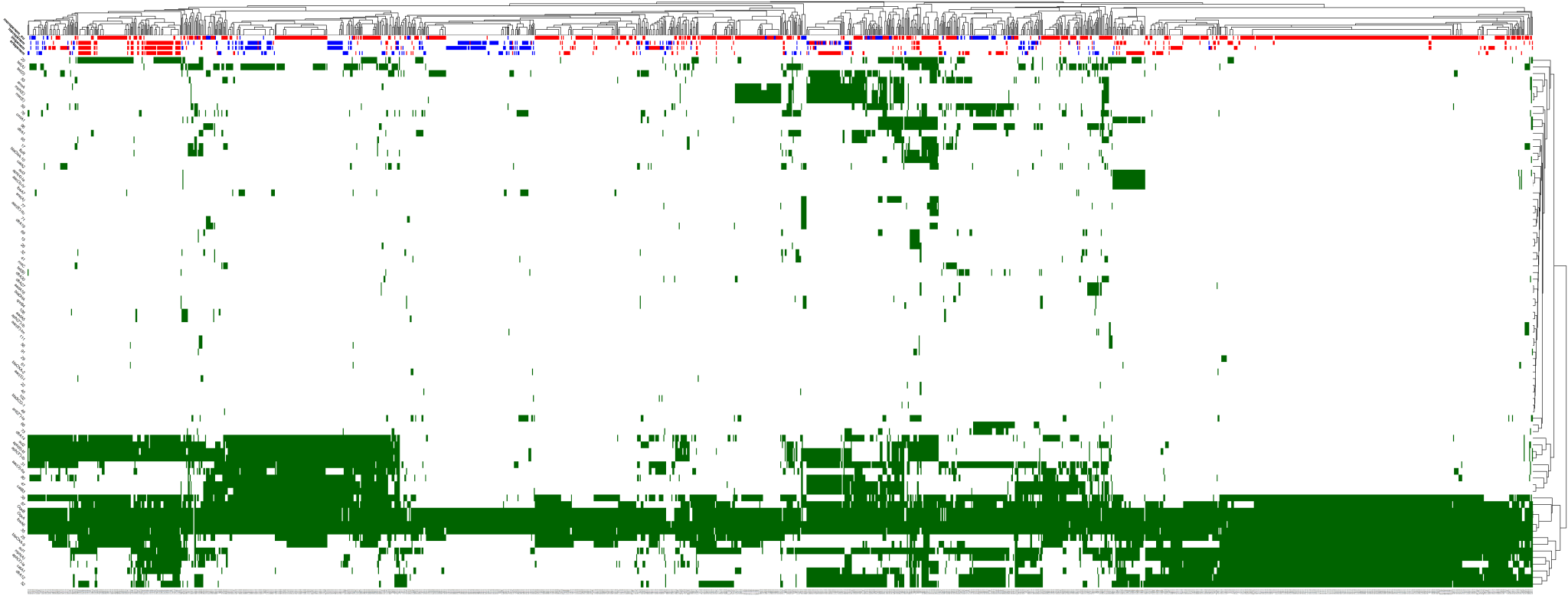
Figure. Distribution of carbapenem resistance genes in bacterial isolates. Analysis and figure by Grete Paat.
2. Development of PCR-based diagnostic tests. For more than 20 years we have studied how DNA molecules hybridize to their specific and non-specific targets. This research is important for designing highly specific PCR primers and microarray probes. We have written several software packages to facilitate genomic PCR primer design. Our workgroup has participated in the development of widely used Primer3 software and we host the official webpage for Primer3, which serves approximately 150,000 users per month.
We have applied our theoretical knowledge in practice by creating various DNA-based molecular tests. Examples include:
* Respiratory disease pathogen detection test for Estonian company Quattromed HTI (now part of SynLab Eesti)
* Blood sepsis-related pathogen tests for the Dutch start-up company Microbiome Ltd (van den Brand et al., 2014)
* Food allergen detection test for the Estonian biotech company Icosagen Ltd. Additionally, we have developed multiplex PCR tests for rapid detection of MLST sequence types of the foodborne pathogen Listeria monocytogenes (Andreson, 2026).
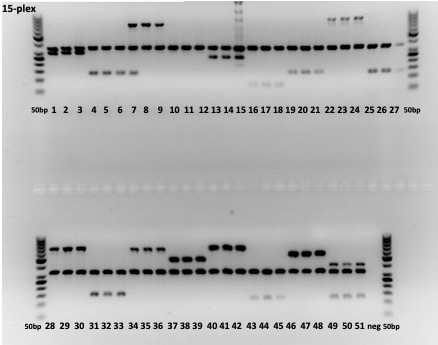
Figure. Multilex-PCR experiment demonstrating the specificity of primers designed for each MLST type (groups of three). The universally present middle lane represents the signal from a universal Listeria-specific primer pair used as a positive control. Data from Andreson et al., 2026.
3. Development of computational methods for metagenomic data analysis. A major advantage of DNA sequencing tests over PCR-based tests is their ability to detect all pathogens simultaneously, whereas PCR can identify only one pathogen per primer pair.
We are currently developing software and databases for reliable detection of pathogenic microbial species and strains from cell-free DNA in human blood samples.
Another active research area is food metagenomics. Notable examples include Raime and Remm, 2018 and Raime et al., 2020, where we described methods and proof-of-principle tests for detecting potentially allergenic food components using a metagenomic sequencing approach.

Figure. Cookies with varying lupin flour content. After baking at 200C, their DNA was extracted, sequenced, and analysed. Experiment and photo by Dr. Kairi Raime.
4. Development of fast and innovative computational methods for personal genome analysis. Over the past decade, we have developed several novel and efficient methods for detecting single nucleotide variants (SNVs) in human personal genomes. FastGT (Pajuste et al., 2017) determines the genotypes of all previously known SNVs in a given genome in just 30 minutes. KATK identifies all SNVs — both known and novel — in approximately 3 hours (Kaplinski et al., 2021).
In addition, we design algorithms for detecting structural variants, which are often overlooked in GWAS analyses. For example, our methods can identify polymorphic Alu-element insertion sites (Puurand et al., 2019), gene copy numbers (Pajuste et al.,2023) and copy number variations in tandem repeats, such as those located upstream of ACAN, MAO and TRIB3 genes (Örd et al., 2020).
Our algorithms employ an original k-mer based approach to genomic sequence analysis, enabling us to process human personal genome data ca 30x faster than traditional methods, without compromising accuracy.
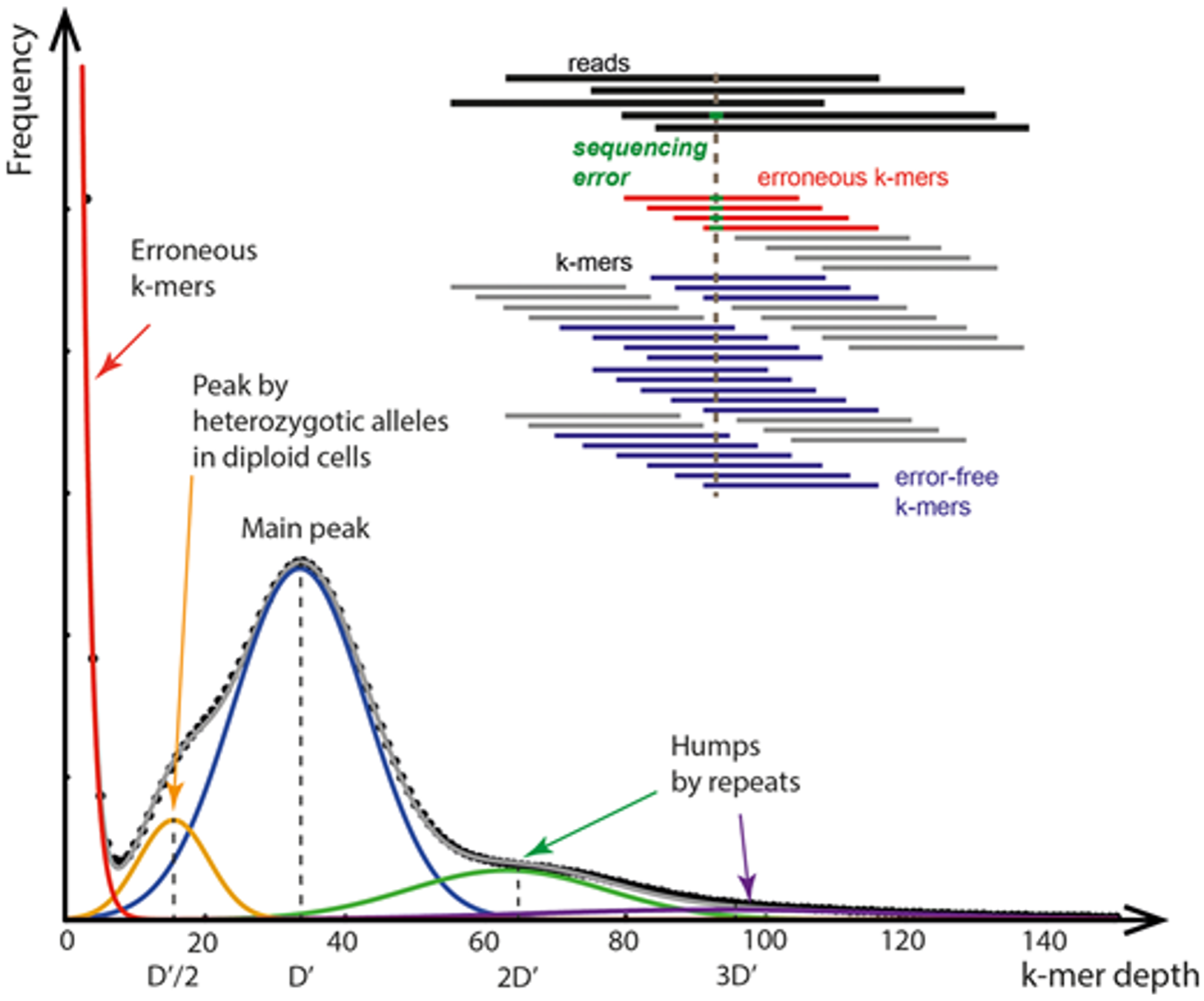
Figure. Typical distribution of unique k-mer frequencies with respect to sequencing depth. Figure created by Tarmo Puurand and Märt Möls